

Hypersensibilité à la substance active ou à l’un des excipients.
Infection active et cliniquement importante (par exemple une tuberculose active ; voir rubrique : Mises en garde spéciales et précautions d’emploi).
Afin d’améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
L’ustékinumab peut potentiellement augmenter le risque d’infections et réactiver des infections latentes. Lors des études cliniques et dans une étude observationnelle après mise sur le marché chez des patients atteints de psoriasis, de graves infections bactériennes, fongiques et virales ont été observées chez des patients recevant l’ustékinumab (voir la liste des effets indésirables). Des infections opportunistes, notamment une réactivation de la tuberculose, d’autres infections bactériennes opportunistes (dont infection mycobactérienne atypique, méningite à listeria, pneumonie à legionella, et nocardiose), des infections fongiques opportunistes, des infections virales opportunistes (dont encéphalite causée par Herpès simplex 2), et des infections parasitaires (dont toxoplasmose oculaire) ont été rapportées chez les patients traités par ustékinumab.
Des précautions doivent être prises lorsque l’utilisation de SteQeyma® est envisagée chez les patients présentant une infection chronique ou ayant des antécédents d’infections récurrentes (voir rubrique : Contre-indications).
Avant d’initier le traitement par SteQeyma®, les patients doivent être examinés pour dépister une infection tuberculeuse. SteQeyma® ne doit pas être administré à des patients présentant une tuberculose active (voir rubrique : Contre-indications). Le traitement d’une tuberculose latente doit être initié avant l’administration de SteQeyma®. Un traitement anti-tuberculeux doit également être envisagé avant l’initiation de SteQeyma® chez les patients ayant des antécédents de tuberculose latente ou active pour lesquels le suivi d’un traitement adapté ne peut être confirmé. Les patients recevant SteQeyma® doivent être étroitement surveillés pour dépister les signes et symptômes de tuberculose active pendant et après le traitement.
Les patients doivent être informés de la nécessité de consulter un médecin en cas de survenue de signes ou symptômes évocateurs d’une infection. Si un patient développe une infection grave, le patient devra être étroitement surveillé et SteQeyma® ne devra pas être administré avant la guérison de l’infection.
Les immunosuppresseurs comme l’ustékinumab sont susceptibles d’augmenter le risque de tumeurs malignes. Certains patients ayant reçu l’ustékinumab au cours d’études cliniques et des patients atteints de psoriasis dans une étude observationnelle après mise sur le marché ont développé des tumeurs malignes cutanées et non-cutanées (voir rubrique : Effets indésirables). Le risque de tumeur maligne peut être supérieur chez les patients atteints de psoriasis qui ont été traités avec d’autres médicaments biologiques au cours de leur maladie.
Aucune étude n’a été conduite chez des patients avec antécédents de tumeur maligne ou chez des patients ayant continué leur traitement alors qu’ils avaient développé une tumeur maligne sous ustékinumab. En conséquence, des précautions doivent être prises lorsqu’un traitement par SteQeyma® chez ces patients est envisagé.
Tous les patients doivent être surveillés pour éviter l’apparition d’un cancer de la peau en particulier ceux âgés de plus de 60 ans, ceux avec des antécédents médicaux de traitements prolongés par immunosuppresseurs ou ceux ayant des antécédents de traitement par puvathérapie (voir rubrique : Effets indésirables).
Systémique
Des réactions d’hypersensibilité graves ont été rapportées depuis la mise sur le marché du produit, dans certains cas plusieurs jours après le traitement. Des anaphylaxies et angioœdèmes sont survenus. En cas de survenue d’une réaction anaphylactique ou d’une autre réaction d’hypersensibilité grave, un traitement approprié doit être instauré et l’administration de SteQeyma® doit être interrompue (voir rubrique : Effets indésirables).
Réactions liées à la perfusion
Des réactions liées à la perfusion ont été observées dans des études cliniques (voir rubrique : Effets indésirables). De graves réactions liées à la perfusion, notamment des réactions anaphylactiques, ont été rapportées depuis la mise sur le marché du produit. En cas de survenue d’une réaction grave ou mettant en jeu le pronostic vital, un traitement approprié doit être instauré et l’administration de l’ustékinumab doit être interrompue.
Respiratoire
Des cas d’alvéolite allergique, de pneumopathie à éosinophiles et de pneumopathie organisée non infectieuse ont été rapportés depuis la mise sur le marché d’ustékinumab. Les symptômes cliniques comprenaient toux, dyspnée, infiltrats interstitiels après une à trois administrations. Les complications graves incluaient insuffisance respiratoire et prolongation d’hospitalisation. Une amélioration a été rapportée après arrêt de l’ustékinumab et aussi dans certains cas après administration de corticoïdes. Si l’infection a été exclue et le diagnostic confirmé, l’administration de l’ustékinumab doit être interrompue et un traitement approprié doit être instauré (voir rubrique : Effets indésirables).
Des événements cardiovasculaires dont infarctus du myocarde et accident vasculaire cérébral ont été observés chez des patients atteints de psoriasis exposés à l’ustékinumab dans une étude observationnelle après mise sur le marché. Les facteurs de risque de maladie cardiovasculaire doivent être régulièrement évalués au cours du traitement par SteQeyma®.
Il est recommandé que les vaccins vivants viraux ou bactériens (tels que Bacille de Calmette- Guérin (BCG)) ne soient pas administrés de manière concomitante à SteQeyma®. Aucune étude spécifique n’a été conduite chez des patients qui ont reçu récemment un traitement par un vaccin vivant viral ou bactérien. Aucune donnée n’est disponible sur la transmission secondaire d’infection par vaccins vivants chez les patients recevant l’ustékinumab. Avant toute vaccination par un traitement vivant viral ou bactérien, le traitement par SteQeyma® doit être arrêté au moins 15 semaines avant et peut être repris au moins 2 semaines après la vaccination. Les prescripteurs doivent consulter le Résumé des Caractéristiques du Produit des vaccins spécifiques pour toute information supplémentaire ou recommandations sur l’utilisation concomitante d’agents immunosuppresseurs après vaccination.
L’administration de vaccins vivants (tels que le vaccin BCG) aux nourrissons exposés à l’ustékinumab in utero n’est pas recommandée au cours des douze mois suivant la naissance ou jusqu’à ce que les taux sériques d’ustékinumab soient indétectables chez le nourrisson (voir les rubriques : Interactions avec d’autres médicaments et autres formes d’interactions et Fertilité, grossesse et allaitement). En cas de bénéfice clinique évident pour le nourrisson, l’administration d’un vaccin vivant peut être envisagée de manière plus précoce si les taux sériques d’ustékinumab chez le nourrisson sont indétectables.
Les patients recevant SteQeyma® peuvent recevoir de façon concomitante des vaccins inactivés ou non vivants.
Le traitement au long cours par ustékinumab ne réprime pas la réponse immunitaire humorale aux vaccins pneumococciques polysaccharidique et antitétanique (voir rubrique : Propriétés pharmacodynamiques).
Dans les études sur le psoriasis, la sécurité et l’efficacité de l’ustékinumab en association avec des immunosuppresseurs, y compris des traitements biologiques, ou de la photothérapie n’ont pas été évaluées. Dans les études sur le rhumatisme psoriasique, il n’est pas apparu que l’utilisation concomitante de méthotrexate (MTX) influence la sécurité ou l’efficacité de l’ustékinumab. Dans les études sur la maladie de Crohn et la rectocolite hémorragique, l’utilisation concomitante d’agents immunosuppresseurs ou de corticoïdes n’a pas semblé influencer la sécurité ou l’efficacité de l’ustékinumab. Des précautions doivent être prises avant l’utilisation concomitante d’autres immunosuppresseurs avec SteQeyma® ou lors du relais après d’autres immunosuppresseurs biologiques (voir rubrique : Interactions avec d’autres médicaments et autres formes d’interactions).
L’ustékinumab n’a pas été évalué chez des patients qui ont été désensibilisés pour leur allergie. On ne sait pas si l’ustékinumab peut interférer avec un traitement de désensibilisation pour allergie.
Chez les patients atteints de psoriasis, une érythrodermie a été rapportée à la suite d’un traitement par ustékinumab (voir rubrique : Effets indésirables). Dans le cadre de l’évolution naturelle de leur maladie, les patients atteints de psoriasis en plaques peuvent développer un psoriasis érythrodermique avec des symptômes pouvant être cliniquement indifférenciables d’une érythrodermie. Dans le cadre du suivi des patients atteints de psoriasis, les médecins doivent être vigilants en cas de symptômes de psoriasis érythrodermique ou d’érythrodermie. Si ces symptômes apparaissent, un traitement approprié doit être instauré. SteQeyma® doit être arrêté en cas de suspicion de réaction médicamenteuse.
Des cas d’affections liées au lupus ont été rapportés chez des patients traités par ustékinumab, notamment un lupus érythémateux cutané et un syndrome de type lupus. Si des lésions apparaissent, en particulier sur des zones de peau exposées au soleil ou si elles sont accompagnées d’arthralgies, le patient doit immédiatement consulter un médecin. Si le diagnostic d’une affection liée au lupus est confirmé, l’ustékinumab doit être arrêté et un traitement approprié initié.
Sujets âgés (≥ 65 ans)
Chez les sujets âgés de 65 ans et plus ayant reçu l’ustékinumab, aucune différence globale concernant l’efficacité et la sécurité n’a été observée en comparaison avec les sujets plus jeunes dans le cadre d’études cliniques dans les indications approuvées, cependant le nombre de patients âgés de 65 ans et plus n’est pas suffisant pour déterminer s’ils répondent différemment des patients plus jeunes. De façon générale, en raison d’une incidence plus élevée d’infections dans la population âgée, la prudence est recommandée pendant le traitement des sujets âgés.
SteQeyma® contient moins d’1 mmol (23 mg) de sodium par dose, c’est-à-dire essentiellement « sans sodium ». SteQeyma® est cependant dilué avec une solution pour perfusion de chlorure de sodium à 9 mg/mL (0,9 %). Cela doit être pris en compte chez les patients suivant un régime hyposodé (voir rubrique : Précautions particulières d’élimination et manipulation).
SteQeyma® 130 mg contient 10,37 mg de polysorbate 80 (E433) par dose équivalent à 0,40 mg/mL. SteQeyma® contient 0,04 mg (90 mg/1,0 mL) ou 0,02 mg (45 mg/0,5 mL) de polysorbate 80 (E433) par dose équivalent à 0,04 mg/mL. Les polysorbates peuvent provoquer des réactions allergiques.
Les vaccins vivants ne doivent pas être donnés de manière concomitante avec SteQeyma®. L’administration de vaccins vivants (tels que le vaccin BCG) aux nourrissons exposés à l’ustékinumab in utero n’est pas recommandée au cours des douze mois suivant la naissance ou jusqu’à ce que les taux sériques d’ustékinumab soient indétectables chez le nourrisson (voir rubriques : Mises en garde spéciales et précautions d’emploi et Fertilité, grossesse et allaitement). En cas de bénéfice clinique évident pour le nourrisson, l’administration d’un vaccin vivant peut être envisagée de manière plus précoce si les taux sériques d’ustékinumab chez le nourrisson sont indétectables.
Dans les analyses pharmacocinétiques de population des études de phase 3, l’effet des médicaments les plus souvent utilisés de façon concomitante chez des patients présentant un psoriasis (incluant paracétamol, ibuprofène, acide acétylsalicylique, metformine, atorvastatine, lévothyroxine) sur la pharmacocinétique de l’ustékinumab a été exploré. Il n’y a pas eu d’élément suggérant une interaction avec ces médicaments co-administrés. Cette analyse est fondée sur l’observation d’au moins 100 patients (> 5 % de la population étudiée) traités concomitamment par ces médicaments pendant au moins 90 % de la période étudiée. La pharmacocinétique de l’ustékinumab n’a pas été modifiée par l’utilisation concomitante de MTX*, d’AINS**, de 6-mercaptopurine, d’azathioprine et de corticoïdes oraux chez les patients atteints de rhumatisme psoriasique, de la maladie de Crohn ou de rectocolite hémorragique, ou par une exposition préalable à des agents anti-TNFα chez les patients atteints de rhumatisme psoriasique ou de la maladie de Crohn, ou par une exposition préalable à des agents biologiques (c’est-à-dire des agents anti-TNFα*** et/ou le vedolizumab) chez des patients atteints de rectocolite hémorragique. Les résultats d’une étude in vitro et d’une étude de phase 1 chez des sujets atteints de maladie de Crohn active ne suggèrent pas qu’il soit nécessaire d’ajuster la posologie chez les patients recevant de manière concomitante des substrats du CYP450**** (voir rubrique : Propriétés pharmacocinétiques).
Dans les études sur le psoriasis, la sécurité et l’efficacité de l’ustékinumab en association avec des immunosuppresseurs y compris traitements biologiques, ou la photothérapie n’ont pas été évaluées. Lors des études cliniques conduites chez les patients atteints de rhumatisme psoriasique, il n’est pas apparu que l’utilisation concomitante de MTX influence la sécurité ou l’efficacité de l’ustékinumab. Dans les études sur la maladie de Crohn et la rectocolite hémorragique, l’utilisation concomitante d’agents immunosuppresseurs ou de corticoïdes n’a pas semblé influencer la sécurité ou l’efficacité de l’ustékinumab (voir rubrique : Mises en garde spéciales et précautions d’emploi).
Les femmes en âge de procréer doivent utiliser une méthode contraceptive efficace pendant le traitement et au moins pendant les 15 semaines qui suivent l’arrêt du traitement.
Les données, recueillies de manière prospective après exposition à ustékinumab, issues d’un nombre modéré de grossesses avec une évolution connue, incluant plus de 450 grossesses exposées au cours du premier trimestre, n’indiquent pas de risque accru de malformations congénitales majeures chez le nouveau-né. Les études effectuées chez l’animal n’ont pas mis en évidence d’effets délétères directs ou indirects sur la gestation, le développement embryonnaire/fœtal, la parturition ou le développement post-natal (voir rubrique : Données de sécurité préclinique). Cependant, l’expérience clinique disponible est limitée. Par mesure de précaution, il est préférable d’éviter l’utilisation de SteQeyma® pendant la grossesse.
L’ustékinumab traverse la barrière placentaire, il a été détecté dans le sérum de nourrissons nés de patientes traitées par ustékinumab au cours de la grossesse. L’impact clinique en est inconnu, toutefois, le risque d’infection des nourrissons exposés à l’ustékinumab in utero peut être augmenté après la naissance.
L’administration de vaccins vivants (tels que le vaccin BCG) aux nourrissons exposés à l’ustékinumab in utero n’est pas recommandée au cours des douze mois suivant la naissance ou jusqu’à ce que les taux sériques d’ustékinumab soient indétectables chez le nourrisson (voir rubriques : Mises en garde spéciales et précautions d’emploi et Interactions avec d’autres médicaments et autres formes d’interactions). En cas de bénéfice clinique évident pour le nourrisson, l’administration d’un vaccin vivant peut être envisagée de manière plus précoce si les taux sériques d’ustékinumab chez le nourrisson sont indétectables.
Les données issues de la littérature sont limitées et suggèrent que l’ustékinumab est excrété en très faible quantité dans le lait maternel humain. On ne sait pas si l’ustékinumab passe dans la circulation systémique après ingestion. À cause du risque potentiel d’effets indésirables de l’ustékinumab chez les nourrissons allaités, l’arrêt de l’allaitement pendant le traitement et pendant les 15 semaines qui suivent l’arrêt du traitement par SteQeyma® doit être évalué, en tenant compte du bénéfice de l’allaitement pour l’enfant et de celui du traitement par SteQeyma® pour la femme.
L’effet de l’ustékinumab sur la fertilité humaine n’a pas été évalué (voir rubrique : Données de sécurité préclinique).
Les effets indésirables les plus fréquents (> 5 %) dans les phases contrôlées des études cliniques conduites avec l’ustékinumab chez les patients adultes atteints de psoriasis, de rhumatisme psoriasique, de la maladie de Crohn et de rectocolite hémorragique étaient des rhinopharyngites et des céphalées. La plupart ont été considérés comme étant légers et n’ont pas nécessité d’interruption du traitement étudié. Les effets indésirables les plus graves rapportés avec l’ustékinumab sont des réactions d’hypersensibilité graves incluant l’anaphylaxie (voir rubrique : Mises en garde spéciales et précautions d’emploi). Le profil de sécurité global était similaire pour les patients atteints de psoriasis, de rhumatisme psoriasique, de la maladie de Crohn et de rectocolite hémorragique.
Les données de sécurité décrites ci-dessous reflètent l’exposition de sujets adultes à l’ustékinumab dans 14 études de phase 2 et de phase 3 menées chez 6 710 patients (4 135 atteints de psoriasis et/ou de rhumatisme psoriasique, 1 749 atteints de maladie de Crohn et 826 patients atteints de rectocolite hémorragique). Cela inclut l’exposition à l’ustékinumab dans les phases contrôlées et non contrôlées des études cliniques chez les patients atteints de psoriasis, de rhumatisme psoriasique, de maladie de Crohn ou de rectocolite hémorragique pendant au moins 6 mois (4 577 patients) ou au moins 1 an (3 648 patients). 2 194 patients atteints de psoriasis, de maladie de Crohn ou de rectocolite hémorragique, ont été exposés pendant au moins 4 ans tandis que 1 148 patients atteints de psoriasis ou de maladie de Crohn ont été exposés pendant au moins 5 ans.
Liste des effets indésirables très fréquents (≥ 1/10) et fréquents (≥ 1/100 à < 1/10)
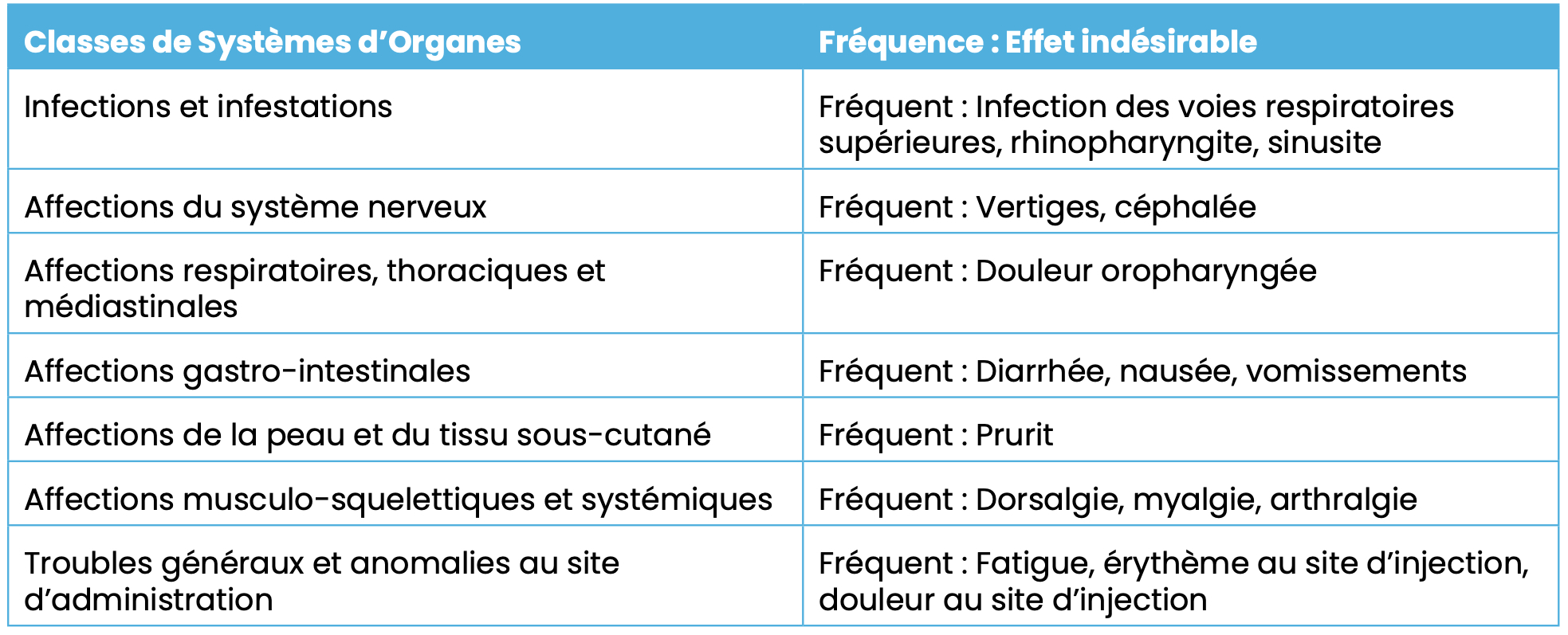
Pour plus d’information sur les effets indésirables peu fréquents (≥1/1 000, <1/100), rares (≥1/10 000, <1/1 000) ou très rares (<1/10 000), consultez le RCP.
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres régionaux de Pharmacovigilance – Site internet : https://signalement.social-sante.gouv.fr/
Des doses uniques allant jusqu’à 6 mg/kg ont été administrées par voie intraveineuse au cours des études cliniques sans toxicité dose-limitante. En cas de surdosage, il est recommandé que le patient soit surveillé afin de détecter tout signe ou symptôme évoquant des effets indésirables et qu’un traitement symptomatique approprié soit immédiatement instauré.
Liste I. Uniquement sur ordonnance.
Médicament biosimilaire. Médicament d’exception.
Médicament réservé à l’usage hospitalier. Prescription réservée aux spécialistes en gastro-entérologie et hépatologie ou en médecine interne.
Agréé Coll. Remb. Séc. Soc. à 65 % dans les indications suivantes :
* MTX : méthotrexate
** AINS : anti-inflammatoire non stéroïdiens
*** TNF : tumor necrosis factor = facteur de nécrose tumorale
**** CYP450 : cytochrome P450
Médicament d’exception : Prescription en conformité avec la Fiche d’Information Thérapeutique. Médicament soumis à prescription médicale restreinte.
Pour une information complète, consultez le Résumé des Caractéristiques du Produit sur la base de données publique du médicament directement sur le site internet : https://base-donnees-publique.medicaments.gouv.fr
![]()
CELLTRION HEALTHCARE FRANCE S.A.S
9-15, rue Rouget de Lisle
92130 Issy-les-Moulineaux
Information médicale et Pharmacovigilance : Tel : +33 (0)1 71 25 27 00
pharmacovigilance_fr@celltrionhc.com
medinfo_fr@celltrionhc.com
FR-STEQ-25-00002 – 25/07/61621908/PM/002 – Décembre 2025
Mentions légales – Politique de confidentialité – Politique des cookies
Aucun cookie nécessitant le consentement de l’utilisateur n’est déposé sur ce site. Seuls des cookies strictement nécessaires et de fonctionnement sont déposés. Pour en savoir plus, veuillez consulter notre Politique des cookies.
